



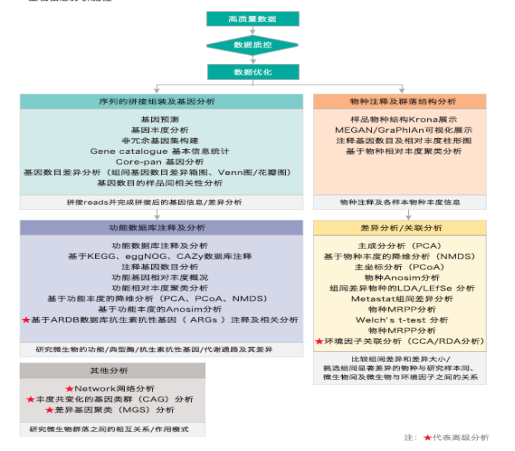
宏基因组数据分析服务
发布时间:2019-04-12 分享到:
数据质控
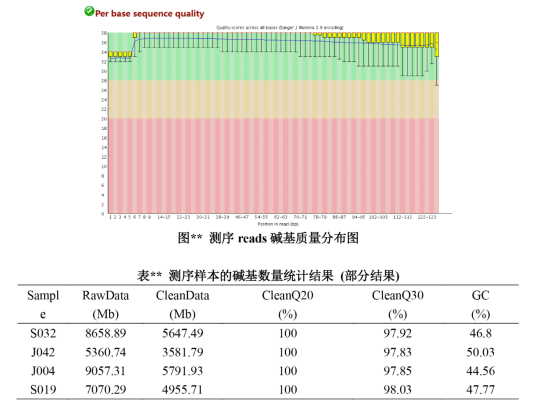
将测序得到的raw reads,需要被剔除接头序列,低质量的序列和嵌合体。如果样品中存在着宿主污染,需要与宿主数据库进行比对,过滤掉来源于宿主的reads。最终得到clean reads。
宏基因组数据组装
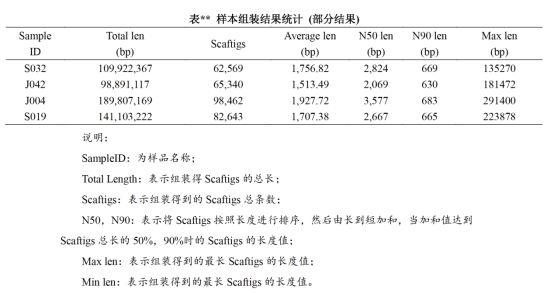
原始下机数据 (Raw Data) 通过质控后获得过滤数据 (clean reads),采用Megahit软件进行Metagenome组装,对组装结果中长度不小于500 bp的scaffolds数据进行统计:
物种组成展示分析
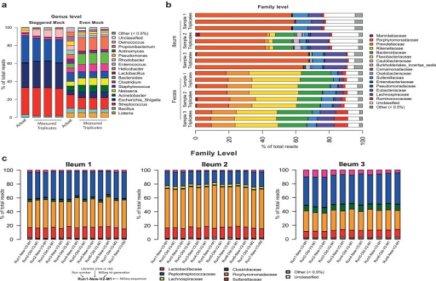
样品菌落结构柱状图(横坐标(Sample Name)是样品名;纵坐标(Relative Abundance)表示相对丰度)
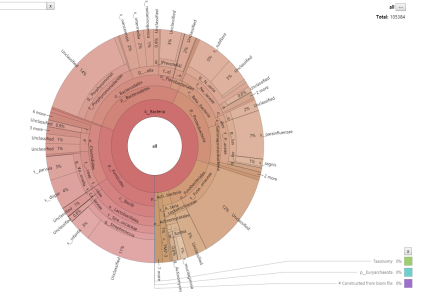
物种Krona注释结果 (圈从内到外依次代表不同的分类级别,扇形的大小代表不同OTU的注释结果相对比例)
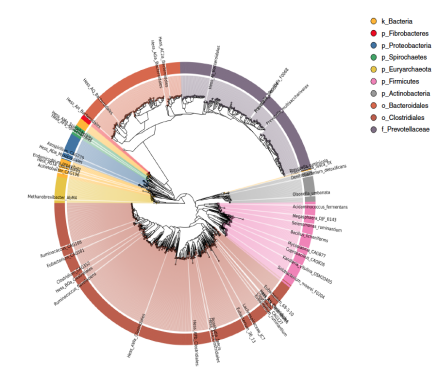
系统进化分支图(在图中可见几个大分支。该树由代表梭菌属和类杆菌属的两个大簇代表,其中后者的一个重要簇代表Prevotellaceae。较小的进化枝代表变形杆菌,古细菌,放线菌,螺旋体和纤维杆菌。其余节点和分支代表杂菌)
基于物种水平的PCA,PCoA和NMDS分析
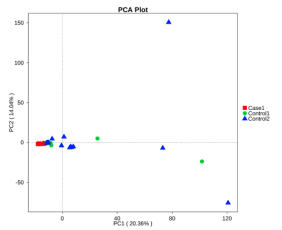
主成分分析图(PCA)
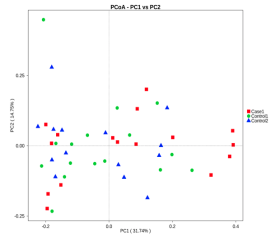
主坐标分析图(PCoA)
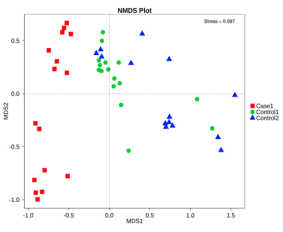
无度量多维标定法(NMDS)
KEGG代谢通路比对
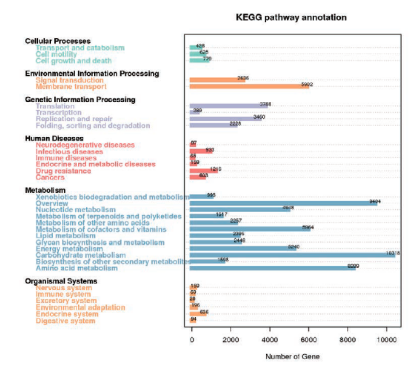
KEGG代谢通路图
CAZyme数据库比对
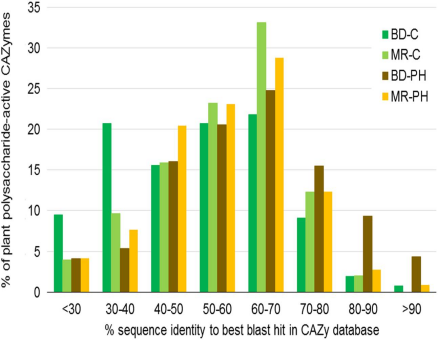
CAZy数据库注释基因数目分析
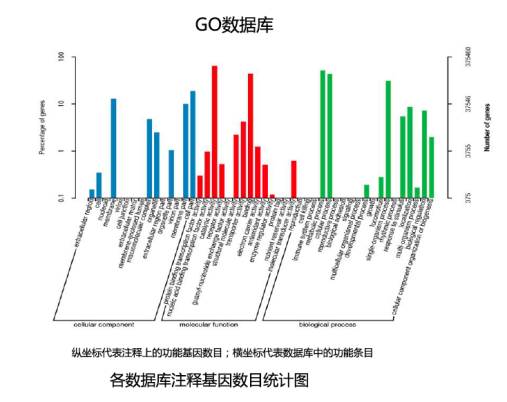
GO数据库分析
相关推荐
- 宏基因组数据分析服务2019-04-12
- 16SrDNA数据分析服务2019-04-12