



GWAS与EWAS联合数据分析服务
发布时间:2019-04-12 分享到:
对于连续型样本性状(Quantitative trait),GWAS通过回归分析,寻找与该性状呈显著关联的SNP位点。这些SNP位点称为“数量性状位点”(Quantitative trait loci;QTL)。当上述连续性状就是甲基化水平(Beta值)时,所得显著关联位点就称为mQTL(Methylation quantitative trait loci)。
mQTL分析
mQTL以分析cis-mQTL及trans-mQTL,前者即利用某基因附近CpG位点甲基化水平Beta值作为因变量,筛选该基因处上下游该区域内的所有SNP变异作为自变量,逐一对该范围内的各个SNP位点与甲基化水平进行回归分析,从而得到与某基因甲基化水平显著相关的SNP位点;后者则针对同一条染色体更远距离(1Mb之外的),或者不同染色体上的SNP位点与CpG位进行线性回归分析,从而得到与某基因甲基化水平显著相关的SNP位点。
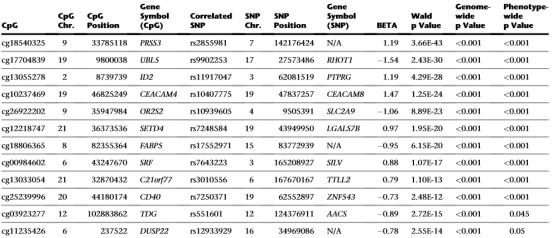
Trans Associations
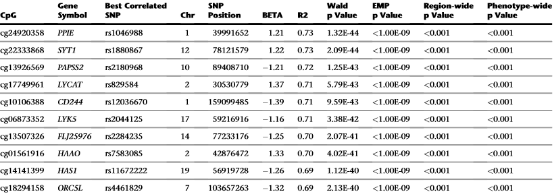
cis Associations
Finemap精细定位分析
通过mQTL初步分析找到的SNP位点往往范围很大,其中很多变异信息仅仅具有统计意义上的关联性,而不是具有因果性。因此产生的候选基因也比较庞杂,包含很多和疾病没有关系的基因。
通过Finemap精细定位分析,进一步缩小易感位点,排除掉仅有关联性,而没有因果性的序列变异。
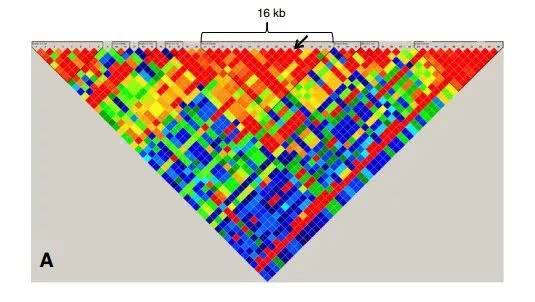
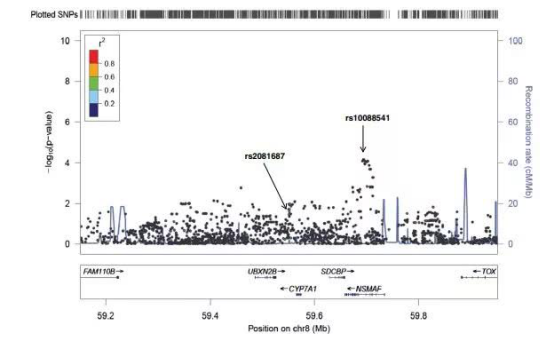
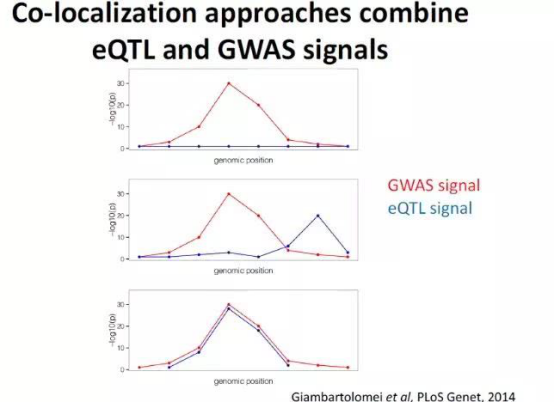
共定位分析
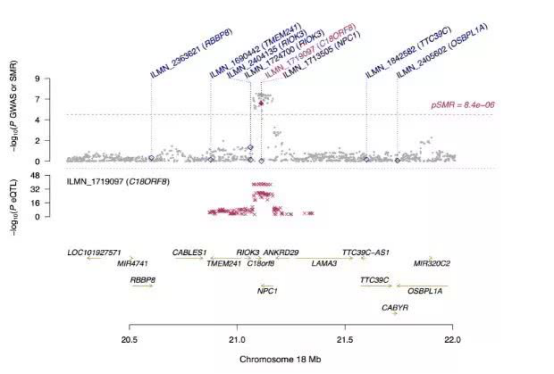
在mQTL分析的基础上,结合GWAS的分析结果,可以进行共定位位点分析, 大部分GWAS显著关联位点都落在基因组非编码区。一种可能的解释是,这些易感位点通过调节特定区域的甲基化水平,从而改变个体复杂性状。如果某个位点既对复杂性状有影响,又对甲基化水平有影响,那么该位点就很有可能符合上述解释。共定位分析(Collocalization)正是试图找出这些“共定位”位点。
SMR利用GWAS的Summary数据和表达数量性状基因座(eQTL)的数据,采用SMR和HEIDI方法,以测试基因表达水平与感兴趣的复杂性状之间的多效性关联。
相关推荐
- GWAS与EWAS联合数据分析服务2019-04-12
- GWAS数据分析服务2019-04-12