



转录组测序数据分析服务
发布时间:2019-04-09 分享到:
数据分析中用到的参考数据包括参考基因组和转录组的序列、基因在基因组上的定位信息,这些参考数据的文件名字、版本以及下载的地址包含在中。另外,分析过程中用到的软件及其版本信息详情见表 2。
|
参考数据 |
来源 |
版本 |
下载地址 |
|
基因组DNA序列文件 |
UCSC |
Homo_sapiens.GRCh37.75.dna.primary_assembly.fa |
ftp.ensembl.org /pub/release-75/fasta/homo_sapiens/dna/ |
|
转录组cDNA序列文件 |
UCSC |
Homo_sapiens.GRCh37.75.cdna.all.fa |
ftp.ensembl.org /pub/release-75/fasta/homo_sapiens/cdna/ |
|
基因定位文件(GTF/GFF) |
UCSC |
Homo_sapiens.GRCh37.75.gtf |
ftp.ensembl.org/ /pub/release-63/gtf/homo_sapiens/ |
|
软件名称* |
来源 |
版本 |
软件说明 |
|
Hisat2 |
CBCB |
2.0.0 |
短序列定位软件 |
|
Stringtie |
CBCB |
2.0.10 |
组装转录本 |
|
rMATS |
rMATS |
3.0.9 |
可变剪切分析 |
|
edgeR |
R |
2.13 |
差异基因分析 |
|
IGV |
IGV |
2.1 |
Reads定位可视化软件 |
|
RseQC |
RseQC |
v2.3.7 |
RNA-seq质量评估 |
|
skewer |
skewer |
0.2.2 |
数据trim方法clean |
|
FastQC |
babraham |
v0.10.0 |
测序质量评估 |
|
ORFfinder |
NCBI |
- |
ORF查找 |
|
FEELnc |
FEELnc |
v0.1.0 |
LncRNA鉴定 |
|
clusterProfiler |
clusterProfiler |
3.1.9 |
功能富集 |
* 网址:
rMATS : http://rnaseq-mats.sourceforge.net/;
clusterProfiler: https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html;
Stringtie:https://ccb.jhu.edu/software/stringtie/;Hisat2:https://ccb.jhu.edu/software/hisat2/index.shtml
IGV:http://www.broadinstitute.org/igv/;RseQC:http://rseqc.sourceforge.net;
skewer:https://sourceforge.net/projects/skewer/
edgeR:http://www.bioconductor.org/packages/2.13/bioc/html/edgeR.html
RseQC:http://rseqc.sourceforge.net/
FEELnc:https://github.com/tderrien/FEELnc
FastQC:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
生物信息学分析内容
1. 对比分析(mRNA和LncRNA)
利用Stringtie软件和人源转录组数据库信息,对样本进行转录本组装;组装完成后再合并所有样本的组装成的转录本;再利用FEELnc软件对合并后的转录本进行LncRNA鉴定;除开LncRNA,其他为mRNA。
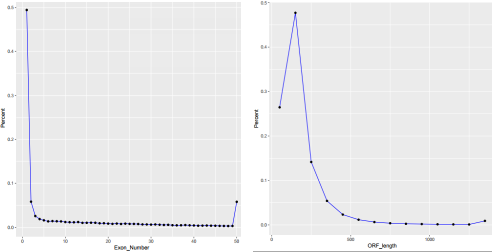
图1:mRNA和LncRNA的exon数目统计和ORF长度分布统计
2. 差异表达分析
通过reads的基因组定位信息再结合mRNA的注释信息,我们可以统计出每个基因上的reads count计数。用edgeR统计组间表达基因的数目;且取FDR<0.05,表达差异2倍以上的基因作为显著差异表达基因。
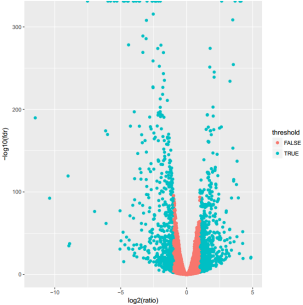
图2:差异表达基因分布的volcano图
表 3. 基因表达差异分析结果(例)
|
Gene |
logFC |
logCPM |
PValue |
FDR |
R1 _ctrl |
R1 _case |
|
CCK |
-11.99366802 |
5.792575182 |
5.76E-23 |
1.32E-18 |
2521 |
0.5 |
|
CST1 |
-9.420238936 |
6.918604601 |
1.04E-21 |
1.19E-17 |
5500 |
8 |
|
SLC1A6 |
-10.07902407 |
5.643436612 |
1.05E-20 |
8.06E-17 |
2271.5 |
2 |
|
LOC284379 |
-9.098469517 |
5.785675968 |
5.96E-20 |
3.42E-16 |
2505 |
4.5 |
|
BASP1P1 |
-10.64163495 |
4.444288603 |
6.22E-19 |
1.78E-15 |
987.5 |
0.5 |
|
COMP |
-8.279951573 |
6.473015767 |
5.81E-19 |
1.78E-15 |
4030.5 |
13 |
|
LINC01518 |
-12.9732932 |
4.460208998 |
5.53E-19 |
1.78E-15 |
999 |
0 |
|
UNC13C |
-8.338309571 |
6.226595199 |
5.17E-19 |
1.78E-15 |
3397.5 |
10.5 |
|
KLK6 |
-12.66631332 |
4.154503341 |
4.51E-18 |
1.15E-14 |
807.5 |
0 |
|
SERPINA4 |
-9.457850314 |
4.110246694 |
6.59E-17 |
1.42E-13 |
782 |
1 |
|
UCA1 |
-7.164225473 |
8.45636275 |
6.81E-17 |
1.42E-13 |
15888.5 |
112 |
|
FREM2 |
-7.114898645 |
7.520565488 |
1.39E-16 |
2.66E-13 |
8302 |
60.5 |
3. PCA分析和聚类分析
对多变量基因表达数据进行主成分(PCA)分析和聚类分析,以评估组内样本的重复性和不同组样本之间的差异性,以剔除异常样本;并可以很直观反映出不同实验条件下样本差异表达基因的变化情况。
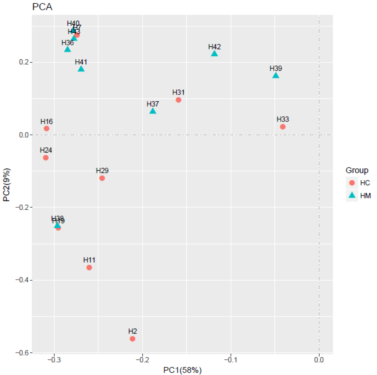
图3:PCA主成分分析
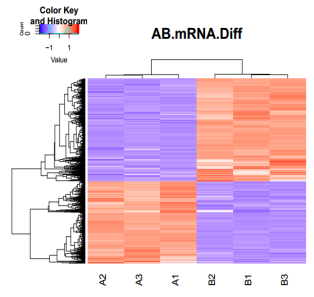
|
图4:聚类分析
4. 基因注释和功能分析
Gene Ontology(简称GO)是一个国际标准化的基因功能分类体系,提供了一套动态更新的标准词汇表(Controlled Vocabulary)来全面描述生物体中基因和基因产物的属性。
GO分析总共有三个Ontology,分别描述基因的分子功能(Molecular Function)、细胞组分(Cellular Component)、生物过程(Biological Process)。采用clusterProfiler注释工具进行GO分析,计算得到的p-value通过校正之后,以q<0.05为阈值,满足此条件的GO条目定义为在差异表达基因中显著富集的GO条目。通过GO功能显著性富集分析能确定差异表达基因行使的主要生物学功能。
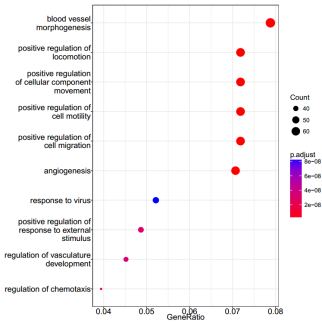
图5:TOP 10的GO富集条目
KEGG(Kyoto Encyclopedia of Genes and Genomes,京都基因与基因组百科全书)是基因组破译方面的数据库。在给出染色体中一套完整基因的情况下,它可以对蛋白质交互(互动)网络在各种各样的细胞活动过程起的作用做出预测。KEGG的PATHWAY数据库整合当前在分子互动网络(比如通路、联合体)的知识,GENES/SSDB/KO数据库提供关于在基因组计划中发现的基因和蛋白质的相关知识,COMPOUND/GLYCAN/REACTION数据库提供生化复合物及反应方面的知识。
其中,基因数据库(GENES Database)含有所有已知的完整基因组和不完整基因组。有细菌、蓝藻、真核生物等生物体的基因序列,如人、小鼠、果蝇、拟南芥等等;通路数据库(PATHWAY Database)储存了基因功能的相关信息,通过图形来表示细胞内的生物学过程,例如代谢、膜运输、信号传导和细胞的生长周期;配体数据库(LIGAND Database)包括了细胞内的化学复合物、酶分子和酶反应的信息。对KEGG中每个Pathway应用超几何检验进行富集分析,找出差异表达基因显著性富集的Pathway。
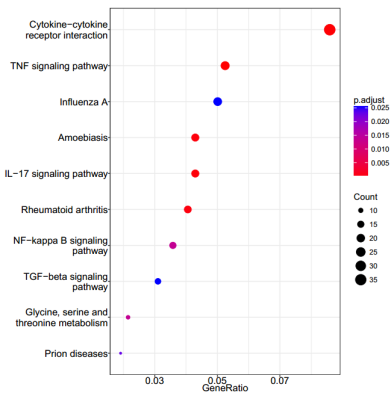
图6:TOP 10的Pathway富集条目
每个点表示该KEGG条目的富集程度,颜色越趋近于红色表示富集程度越高。每个点的大小表示富集到该KEGG条目的基因的个数,点越大表示富集到该KEGG条目的基因越多,反之则越少。
5. 可变剪切分析
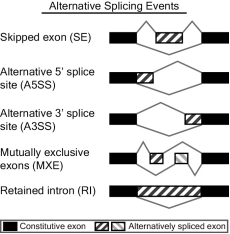
mRNA前体经不同的剪接方式或选择不同的剪接位点将产生多种mRNA剪切异构体,该过程即称为可变剪切(AS)。可变剪切(Florea, et al., 2013)广泛存在于真核生物中,是调节基因表达和蛋白质多样性的重要机制。
|
可变剪切事件的类型整体有以下5类,如下图所示:
图7:5种主要可变剪切的示意图
使用软件rMATS(基于鉴定得到的lncRNA和mRNA转录本)对分组样本进行比较,得到样本间差异可变剪切。
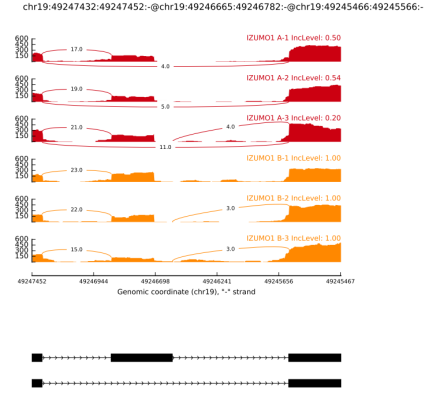
图8:可变剪切示意图
6. LncRNA分析和靶基因预测
对于通过比对分析发现的LncRNA数据,通过预测靶基因(Cis作用靶标和Trans作用靶标)间接预测其功能。
Cis功能预测基本原理。是依据LncRNA的功能与其坐标临近的编码蛋白基因相关。根据找出同LncRNA基因相邻的(上下游20K)蛋白编码的基因对LncRNA筛选。
Trans靶标预测原理,是首先根据LncRNA和基因的表达量计算LncRNA和基因之间的相关性,并且根据相应GTF文件和基因组文件提取对应的Fasta序列,使用RNAPlex根据提取的Fasta序列,计算LncRNA和基因序列之间的结合自由能来预测反式作用靶标。根据自由能和相关性筛选预测靶标,筛选标准Cor>0.75。
相关推荐
- 转录组测序数据分析服务2019-04-09
- miRNA表达谱芯片数据分析服务2019-04-09
- ceRNA数据分析服务2019-04-10